")
")

Bioinformatics
Bio-Fab bioinformatics services are designed to respond to the growing request from our customers for a complete analysis service, from generation of the data to the processing and transformation. Each specific analysis is designed to answer to the needs of the customer who will participate, together with our experts, in the processing of the scientific data produced in the Bio-Fab laboratories. Bio-Fab bioinformatics services are designed to offer advice both pre and post analysis, both on the data obtained by our sequencers and on data delivered directly by the customer. The analyzes, in each of the services and with maximum flexibility, can be basic, advanced and / or customized.
Some Bio-Fab bioinformatics services concern:
- Metagenomic Analysis (Metabarcoding)
- Whole genome assembly (Whole genome seq; plasmid finder; virulence finder ..)
- Differential expression of transcripts (mRNA sequencing)
- Differential expression of miRNAs


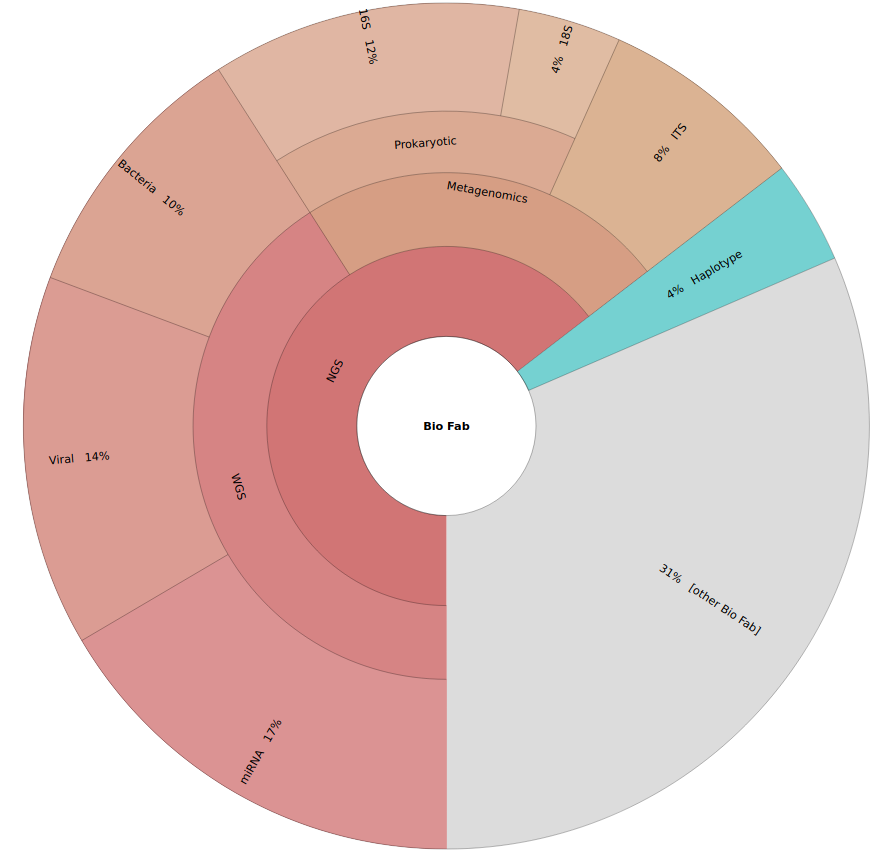
Bio-Fab Services
Word Clouds

COVERAGE: What does it mean in the NGS context?
The term “Coverage” is often used, but sometimes it causes confusion, because in addition to the common definition there are alternative descriptions, various types (including various forms of calculation) and confusion with another term: sequencing depth.
The term "Coverage" in NGS always describes a relationship between reads (sequenced fragments) and a reference (Reference, for example a whole genome, a locus or a location), as opposed to the depth of sequencing which describes the number of total reads. It is important to distinguish between their meanings:
- Coverage in terms of redundancy: number of reads that align or "cover" a reference. It describes how often, on average, each base of a reference sequence is covered by the aligned reads. This is important information because multiple observations of a base at a given location are needed to get a reliable call (for example in variant analyzes). Therefore the coverage in terms of redundancy is also used as a unit for the statistical significance of the sequencing data. Depending on the reference (whole genome or a locus), there are several ways to calculate the coverage.
- Coverage in terms of the percentage of coverage of a reference by the reads aligned with it. For example, if 90% of a reference is covered by reads (and 10% not) we say we have 90% coverage.
- Sequencing depth: total number of fragments sequenced (raw reads) or produced by the sequencing platform (usually expressed in millions of reads).
Among these terms, the only one that can be estimated a priori is the sequencing depth. The estimate of the other two forms of Coverage is dependent on the alignment of the reads to the reference as well as on the quality parameters used for filtering the raw reads during the first steps of bioinformatics analysis. Tutti e tre i significati attribuibili al termine Coverage sono stimati attraverso la stessa formula, riarrangiabile per ciascuna terminologia.
What is good coverage for an NGS project?
There are general or specific guidelines that describe the Optimal Coverage for a sequencing project. It strongly depends on the type of experiment, species, input material, sequencing platform and other factors. However, we recommend that you contact our sequencing service with this question before ordering a project, so that you can leverage the existing experience.
Are there regions in the genome that aren't covered by DNA sequencing?
If the genome were a road and you made a map of the footprints of passersby on that road, the map would certainly have white areas. In other words: some genomic regions cannot be covered very well by sequencing DNA with NGS technology due to repetitive sequences (e.g. tandem repeats) which are abundant across a wide range of species.
About 50% of the human genome is made up of repeats. Repeats are a challenge for sequence assembly and alignment programs especially if short, very similar reads are generated. It can be compared to a large puzzle whereby some puzzle pieces fit together in different places. Hence the repetitions create ambiguities in alignment and assembly, which, in turn, can produce distortions and errors in the interpretation of the results..
The %GC also affects coverage in general. The four bases (ACTGs) are normally not evenly distributed in a genome. Regions of DNA with high and low %GC are difficult to amplify due to higher stability than a DNA region with a mixed base content. In these cases, DNA polymerase is prone to producing artifacts. These effects disturb the amplification steps required in most protocols. As a result, fragments of regions with high / low %GC are underrepresented, with poor and unbalanced coverage. Some current projects try to improve the sequencing of these regions by minimizing the artifacts produced by PCR (amplification in the case of platforms like Illumina) or by giving up the amplification phase (example with MinION).
Finally, fragmentation, in shotgun methods, influences Coverage, because as reported in the literature, DNA fragmentation is a largely non-random process, especially for mechanical cutting methods. This leads to non-uniform Coverage of various genomic regions and can result in regions with zero or low coverage compared to what is estimated / desired.