")
")

FAQs
How are oligos synthesized?
Oligonucleotides are synthesized using a programmable DNA synthesizer basically consisting of a reagent system for the synthesis of oligonucleotides.
The first base is fixed to a solid support, usually a glass or polystyrene ball which anchors the DNA chain forming in the reaction column.
DNA synthesis consists of a series of chemical reactions:
- I phase - Deblocking: the first base, fixed to the solid support by means of a molecular linker, is de-protected by removing a protective Trityl group. This phase generates a 5'OH group that reacts with the next base.
- Phase II - Coupling: the next base is activated and is coupled to the 5'OH group of the last base in the chain.
- III phase - Capping: each base that does not react is protected by a "cap" to prevent it from not taking part in the reaction of the subsequent phases of the synthesis cycle.
- IV phase - Oxidation: the bond between the first base and the second base successfully coupled is oxidized to stabilize the chain in formation.
- V phase - Deblocking: group 5's Trityl is removed from the base to which it was added.
- Each reaction cycle results in the addition of a single DNA base. A DNA base chain can be constructed by repeating several cycles of synthesis until the desired length is obtained.
What is the synthetic scale?
The words “scale synthesis ”refers to the initial amount of controller-pore glass (CPG) monomer bound to the support that is used to initiate DNA synthesis, and not to the amount of final synthesized material. When a scale of 40 nmoles is specified, it means that approximately 40 nmoles of first base are inserted into the synthesizer. For the synthesis of a 25-mer, at least 25% of the starting material will end up in failed sequences. This means that it is not possible to produce 40 nmoles of full-length product from a synthesis scale of 40 nmole. Losses occur during the synthesis, post-synthetic processing, material transfer and quality control phases.
For oligonucleotides with standard length (&alt; = 33 nucleotides) a certain yield of DNA is guaranteed (OD260 applied to 20 mer) which is distributed in a range of +/- 20% relative to the respective synthesis scale and method of purification.
What is coupling efficiency?
The coupling efficiency is used to measure the synthesizer's efficiency in adding new bases to the chain of DNA in formation. If each base available on the chain has successfully reacted with the new base input, the coupling efficiency will be 100%. Only some reactions reach 100% efficiency. During DNA synthesis, the maximum coupling efficiency that can be achieved is normally around 99%. This means that at each pairing step approximately 1% of the available bases are unable to react with the new input base. The coupling efficiency is significantly influenced by the quality of the raw materials used (amidites and solutions), by the conditions of the instrument, and by the synthesis protocol used.
Why is coupling efficiency important?
Coupling efficiency is important because the effects are cumulative during DNA synthesis. Table 1 shows the effects of the coupling efficiency on the percentage of full-length products at the end of the synthesis of oligonucleotides of different length.

Even with a relatively short oligonucleotide of 20 bases, the 1% difference in coupling efficiency can mean an average 15% more DNA present following the synthesis of full-length products.
The table also shows that the longer an oligonucleotide, the lower the expected yield of a full-length product. This phenomenon is due to the type of chemistry used. Even if 99% coupling efficiency is achieved for each addition of a single base, the synthetic raw product of a 95 mer will only consist of 38.5% full-length oligonucleotide.
Separating full-length products for HPLC results in additional loss and therefore lower yields.
What is the tritile group?
Any base added during the synthesis possesses a dimethoxy-trityl protecting group attached to the 5'-hydroxyl position. This acid group protects the base from unwanted chemical reactions during the synthesis cycle and is removed at the first step of each synthesis cycle immediately before adding a new base, until the end of the elongation phase. The final trityl group is either retained or removed, depending on the purification method chosen.
How is the coupling efficiency measured?
The trityl group is colorless when attached to a DNA base but returns a characteristic orange color when removed. The intensity of this color can be measured by means of UV spectrophotometry and is directly related to the number of trityl molecules present. Following the first coupling step, the amount of trityl released during deblocking is directly proportional to the amount of full-length oligonucleotides synthesized in the previous cycle. By comparing the intensities of the trityl produced after the first and last coupling, it is possible to calculate the average base coupling success per cycle and therefore the coupling efficiency.
When I make a large order of oligo often the delivery delays, why?
DNA synthesis is a complicated process that has achieved significant improvement over the past 10 years. Despite this improvement, all manufacturers have a consistent failure rate. We are developing new processes and systems to minimize these failures. However, it is inevitable that we occasionally find ourselves forced to re-synthesize some oligonucleotides.
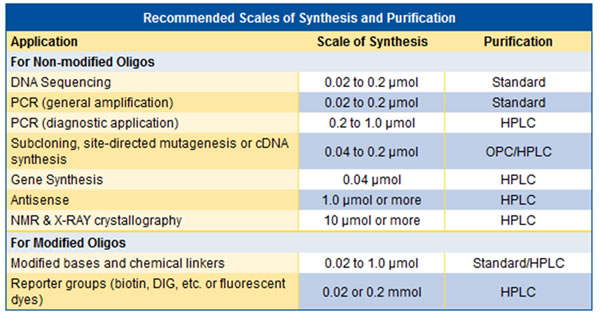
Is it necessary to purify the oligos?
It depends on whether they have been requests for changes or not and from which application will be. Failure sequences can be generated both during the synthesis process and during the post-synthesis process. We recommend that you request that all modifications be purified by HPLC. For the recommended purity and scale, see the table below.
Why do oligos require purifications?
After DNA synthesis, the complete chain is released from the solid support following treatment with a basic solution such as ammonium hydroxide. After the treatment, a new solution is obtained. This contains the required full-length oligonucleotide plus all the DNA chains that are aborted during synthesis. If a 20mer has been synthesized, the solution should also contain aborted 19mer, aborted 18mer, aborted 17mer etc., in small quantities.
The amount of aborted sequences present is influenced by the coupling efficiency and the length of the oligo (Table 1). These aborted sequences can compete with the full-length product in some applications such as sublconing, gene synthesis, antisense. Remember that in general, the higher the degree of purity, the lower the final yield!
Because there are lower yields for modified oligos ?
Many of the modified amides are less stable and do not pair as efficiently as the unmodified bases, so aborted sequences are more abundant than in a normal synthesis. Consequently, most of the modified oligonucleotides should be purified by HPLC to remove the major amount of the aborted sequence. The result is lower yields although the final product is much purer and the probability that the resulting product is totally modified is greater.
Do my oligos have a 5 prime phosphate group?
Unless specifically requested, oligonucleotides are synthesized without 3 'or 5' phosphate.
My oligos annilate but not ligano. Why?
Ligation reactions require a 5 'phosphate. If your oligos do not contain a 5 'phosphate, ligation will not occur or will occur with a very low efficiency. The problem can be solved without ordering another oligonucleotide: enzymatically phosphorylate your oligonucleotides with a kinase before using them for the ligation reactions.
Why isn't there a yield for the 1 um synthesis scale five times higher than the 0.2 um synthesis scale?
For the 0.2, the coupling monomer is made at an excess of moles 40-50 times. Doing it for higher scales of synthesis would be prohibitive in terms of costs. Large-scale syntheses are only done at a 10-fold excess moles of amide and 1 umol columns have not been fully optimized to date in terms of flow, dead space and diffusion. However, to increase the yields of larger synthesis scales, the coupling times are extended to increase coupling efficiencies.
What is the maximum synthesis length for an oligo?
It all depends on the resolving power of the purification method and the coupling efficiency of the synthesizer. We can synthesize 180 base oligos and obtain acceptable quantities for purification in HPLC. In any case, the greater the length of the oligonucleotide, the greater the chances of accumulating sequence errors.
Base composition and synthesis scales are also important factors. Table 1 shows that with 99% coupling efficiency, a 95-mers crude solution should contain 38% of full-length product and 62% of aborted sequences. It is also necessary to take into account the effects of chemical treatments, such as purification. The purification mainly shows effects on the base A. The frequency of purification is low but could significantly increase with the lengthening of the primer.
For these reasons, we indicate 80 bases of maximum length, which we believe to be the length that can be economically synthesized without major problems. Since the likelihood that the synthesis of longer oligos has to be repeated due to premature synthesis breakdown due to low coupling efficiencies, prices for oligonucleotides greater than 80 nucleotides are higher.
What do my oligonucleotides respond to, and at what concentration?
In purified water, TE or any other biological buffer accepted as a diluent. The recommended volume of diluent is between 100 ul and 1 ml, the concentration depends on the application for which the oligos must be used and the yield of the final product. The standard concentration for PCR primers is 0.1 mM.
How stable is my oligonucleotide after I resuspend it?
If a sterile diluent is used, it will be stable at 20 ° C for several days or weeks, at 4 ° C for about a month. If stored at -20 ° C or -70 ° C, it will be stable for several months, even years. The repetition of freezing-thawing must be avoided, otherwise the oligonucleotide will be denatured.
For modified oligonucleotides - especially for those labeled with fluorescent probes - in addition to the above, it would be necessary to minimize exposure to light due to the bleaching effect. We also recommend storing the oligonucleotides labeled with probes at high concentrations and not diluting them if not intended for use within 24 hours. The higher the dilution factor, the faster the fluorescence decay will occur. Furthermore, it is possible to aliquot them, dissolving one aliquot at a time, diluting the aliquot just before use and keeping the dilution in the dark at 4 ° C. If an oligonucleotide is lyophilized and not resuspended, it can be stored unopened at room temperature or indefinitely at -20 ° C. If the oligonucleotide is resuspended, it will be necessary to store it at -20 ° C or at lower temperatures.
Are there any guidelines to follow when designing oligonucletides?
Yes and they are the following:
Sequence length - oligonucleotides from 5 to 180 or more bases are usually synthesized. Most sequences range from 18 to 30 bases with an average of 24 bases. It must be remembered that the longer the oligo, the lower the percentage of full-length product in the raw synthesis leading to a reduction in yields after purification.
Composition of the sequence - it must be ensured that the sequence does not result in hairpin structures. More than 6 consecutive repeated bases (e.g. GGGGGGG) can be problematic as they reduce final yields.
Modifications - Whenever possible, it is necessary to choose modifications to the 5 'terminal. DNA synthesis occurs automatically in the 3 '- > 5 '. Each addition of nucleotide is 98-99% efficient with a percentage of 1-2% of truncated and capped oligo at each position. By placing the modification at the 5 'terminal, it is ensured that only the full length oligonucleotide is modified. Furthermore, since most of the modifications are more hydrophobic than the unmodified oligonucleotide, the modified full-length oligo binds more closely to the reverse phase during purification by HPLC.
Synthesis scale - The term “synthesis scale” refers to the amount of derivatized solid support used. The final amount of product obtained will depend on the length of the sequence, the sequence of the secondary structure, the type of modification chosen, the placement of the modification, the number of modifications per nucleotide and the purification method used.
Purification method - the purification method must be chosen based on the purity level required by the specific application.
Why MALDI-TOF?
In the production procedure of oligo http, each single oligonucleotide is normally analyzed for MALDI TOF to obtain the mass spectrum and compare it to the spectrum of theoretical expected mass. Documentation of the http quality control procedure can be provided upon request at an additional cost.